Time: 2hrs
Difficulty: Moderate
###Objectives
Tutorial 1
We have a protein sequence but we don’t know anything about it. Your task is to try to characterise the protein.
- Download the protein sequence from here:
http://147.188.173.140/public/researcher/mystery_protein.faa
- Try opening the file.
Often files with extensions other that .txt are actually text files. In bioinformatics it is good practice to open text files in something like notepad (for Windows users) and textedit (for Mac users - ensure that format is set to plain text).
-
Your file will look something like this:
>my protein|abc1
MSQLLNDTLSAWLLIESLSPGEVNFTAEDILSAEHFKNGAKQAQLQSFDEYFEIWNSERF
IISEEKSETGELIFKFYRHCFRYNEINLKIQDIFDDYSDIHNPNGTHCYGYTFNTDKHGK
VIVDSIHIPMIMSALKEIEKNKNANIEEKFNDSVEKFFQKVKEILADEPINEFKLKKMDK
AYDEYFSVLNSKKDGLFGHYVAIEYVKDSDLPQPEFNSFFISDIEKARKSPNQTLIDYIE
This is a fasta file. These can come in nucleotide format (.fna .fasta .ffn) or amino acid format like this one (.faa .fasta). Wikipedia actually give a good explanation of this format and what the different extensions mean, here.
True fasta format always starts with line with a > this is the header line. This can contain myriad of things including name, description, accession number, product. The header line is always followed by sequence.
- Some files are multifasta, what do you think this means?
In order to find out what our sequence is we are going to use a piece of software called BLAST (Basic Local Alignment Search Too). As the name says this performs a local alignment. This tool searches against sequence databases very quickly, it is probably the most commonly used tool in biological science.
-
Why do you think that a local alignment is more appropriate than a global alignment when you are blasting a protein against a genome? See here for a succinct explanation of the differences.
-
Go to https://blast.ncbi.nlm.nih.gov/Blast.cgi and select protein BLAST. Either copy and paste mystery_protein.faa into the yellow box or choose the upload option and upload the file and then press submit in the bottom left corner.
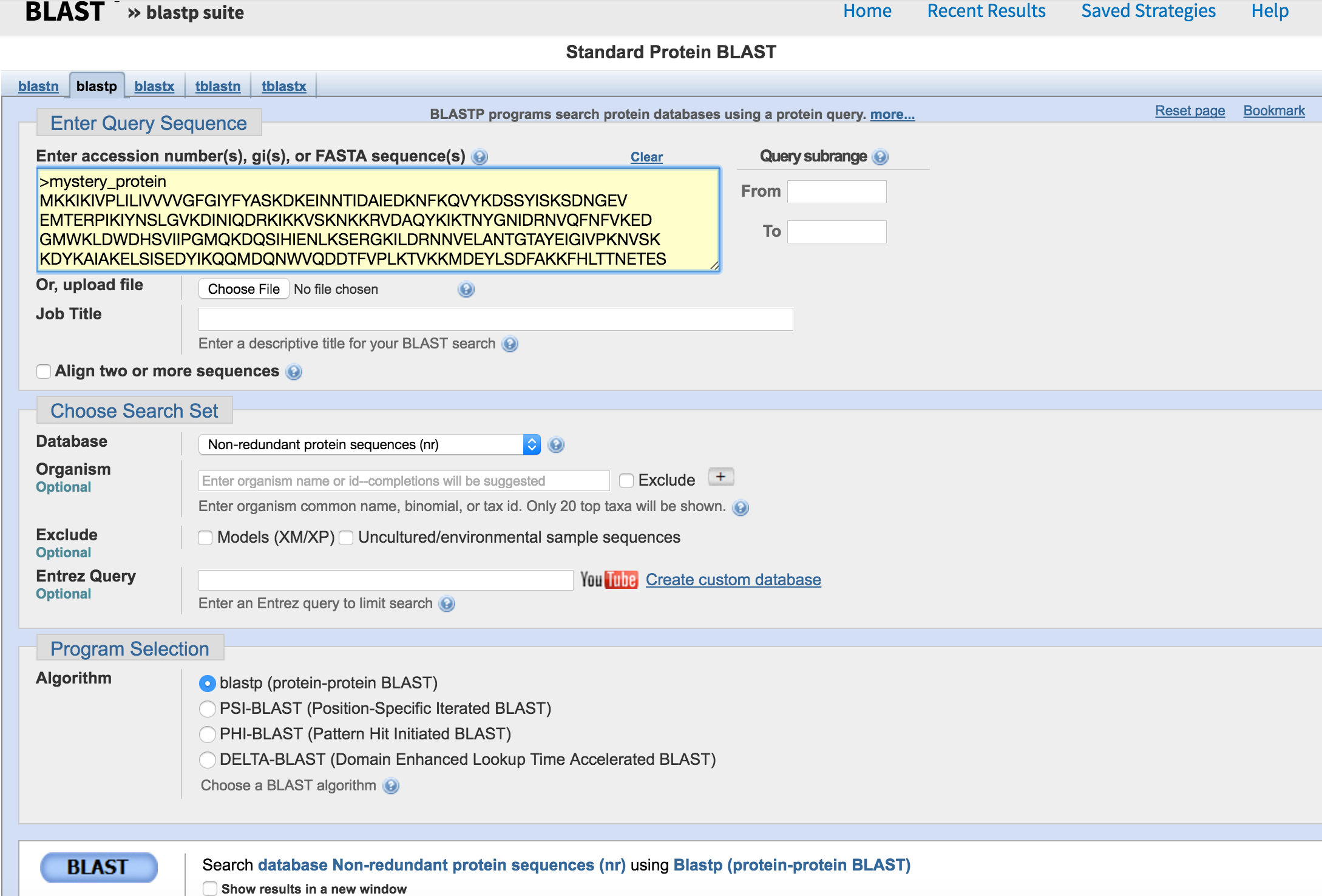
The next two slides explain the blast output that you are seeing and what it means. They are derived from a lecture by Mark Pallen on bacterial genome analysis and annotation here. This is a great general overview.


- What is your mystery protein?
- What organism does it come from?
- Perform the BLAST search again but using tblastn. How are the results different?
Did you know that you can blast against specified organisms?
- Go back to the BLAST front page (you can do this by clicking on edit and resubmit)
- Change the search type to tblastn (tab near the top) and in the Organism text box and enter Staphylococcus aureus subsp. aureus N315 (taxid:158879) rerun this.
- How is blastp different to tblastn?
You’ll notice that you have two hits, we are interested in the complete genome. Click on the hit called select ‘Staphylococcus aureus subsp. aureus N315 DNA, complete genome’
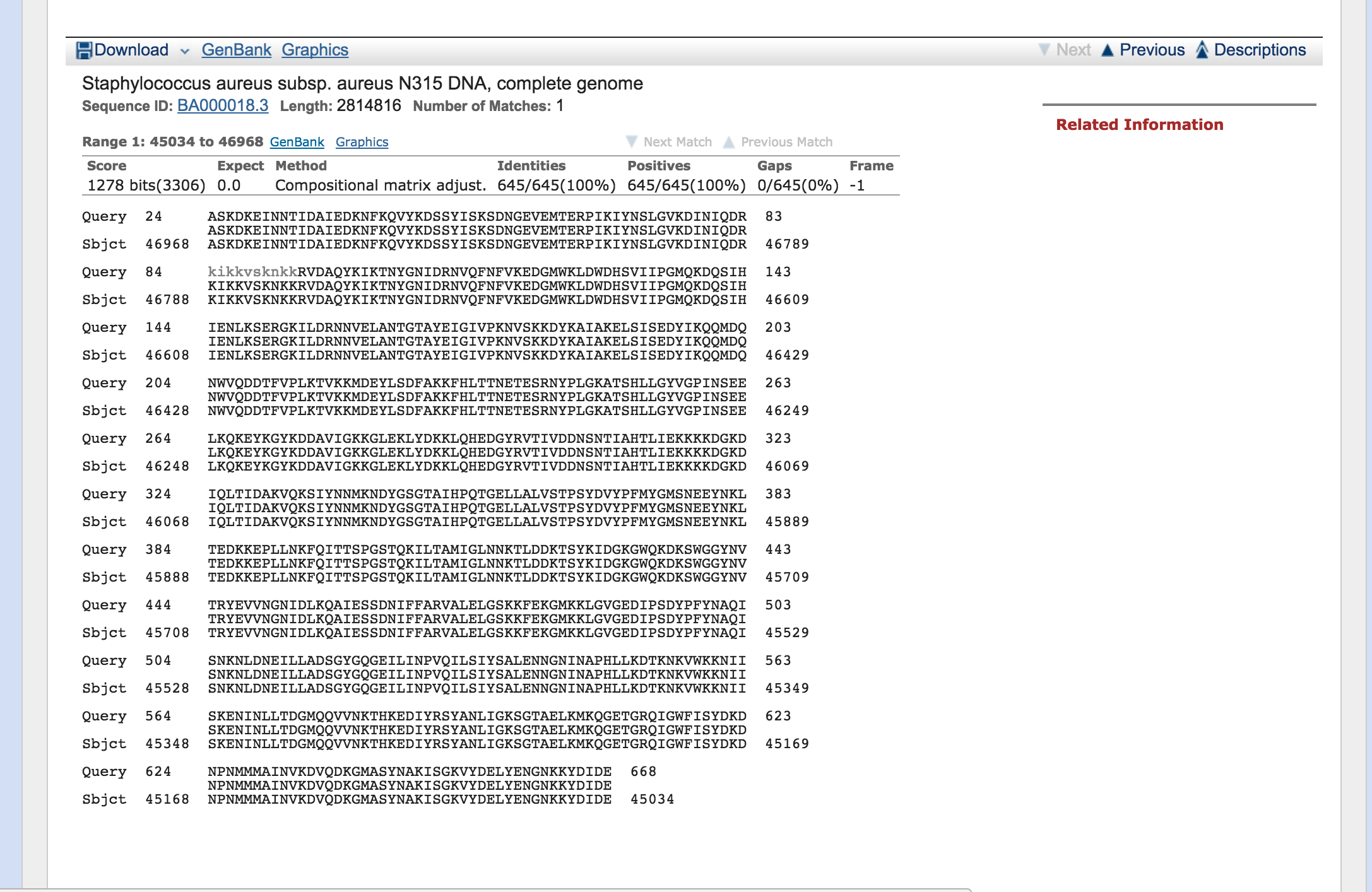
- Now click on the Genbank link

You are now looking at the annotation of the hit in the Staphylococcus aureus N315 genome. This annotation is in genbank format. Annotation formats tend to be human readable. This means that it is easy for a person to read and interpret but normally not readily interpretable by a computer. Genbank format has a header (Title, taxonomy, citation), then features (genes, CDS, RNAs etc.) followed by the nucleotide sequence.
We want to know about the context of our protein. To do this we are going to download the some sequence either side of where the protein is in the N315 genome. The sequence either side of your region of interest is called the flanking region.
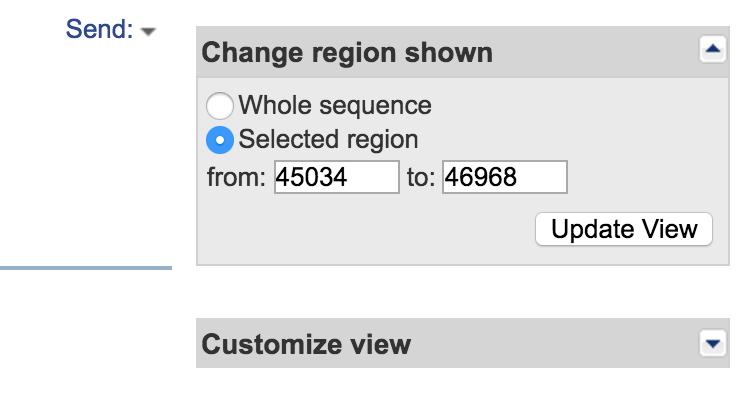
- Change the selected coordinates to 26500 and 97500 (ensure that Show sequence is checked) and click Update View
- When you look at the annotation now you can see some of the genes surrounding your protein. Does it give any clues as to what this protein is. Is it on a mobile genetic element?

From this portal you can actually view the whole genome and download/view it in different formats.
-
Can you download the entire genome for Staphylococcus N315 in genbank format and then fasta format? Name the N315.genbank and N315.fasta respectively
-
Can you use your mystery protein in these databases? What more do they tell you?
http://pfam.xfam.org/
http://www.ebi.ac.uk/interpro/search/sequence-search
http://www.rcsb.org/pdb/home/home.do
http://www.uniprot.org/ #look for the blast tab
Citations #This should be in Uniprot
Gene Ontology terms #This should also be in Uniprot
Now that we know a bit more about this protein we want to compare this region to Staphylococcus aureus ED98. Search the NCBI for this genome and download the entire genbank sequence.
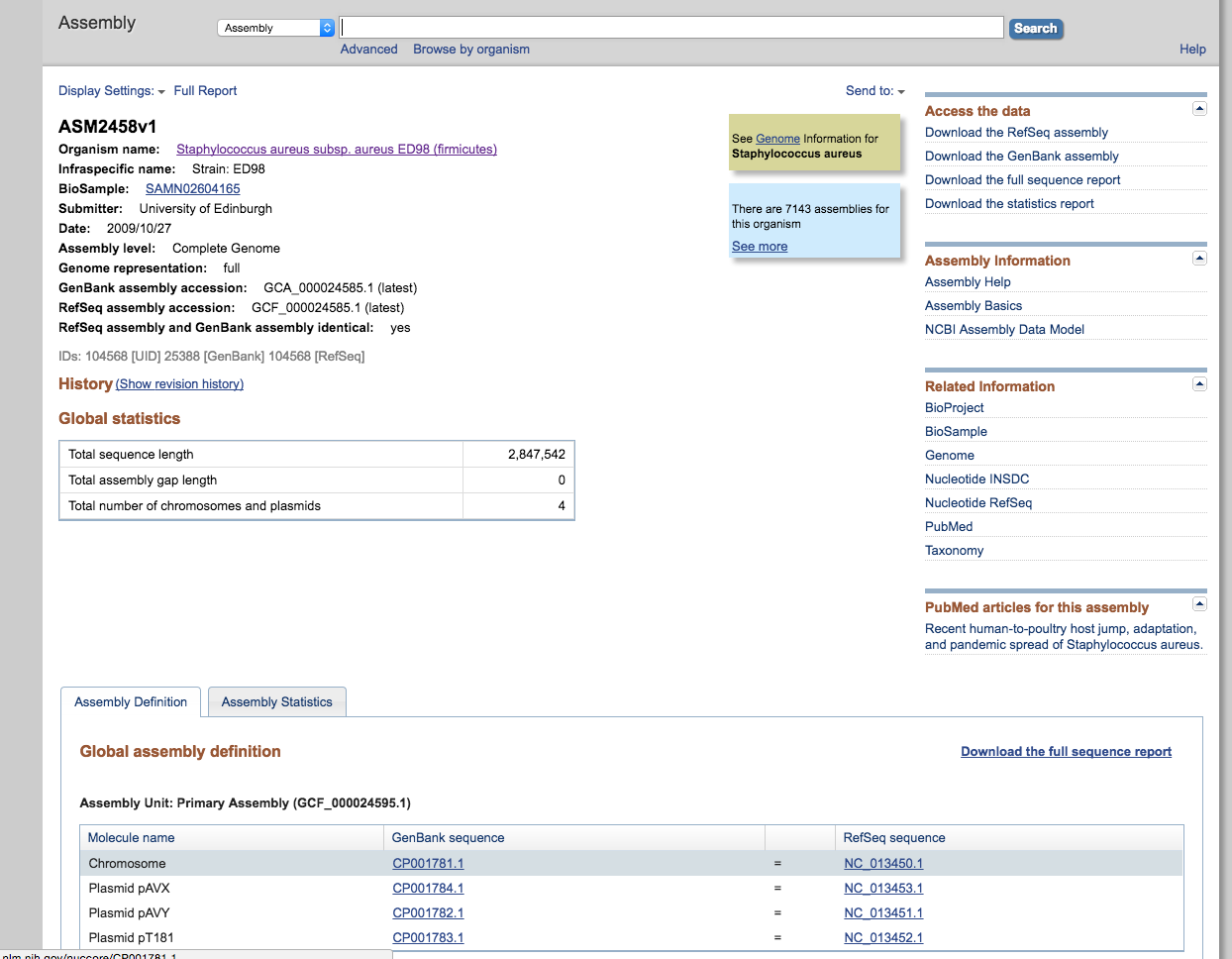
- Select the Chromosome link to the refseq annotation highlighted at the bottom, then download the genbank and fasta file like you did before. Call these ED98.gbk and ED98.fasta respectively
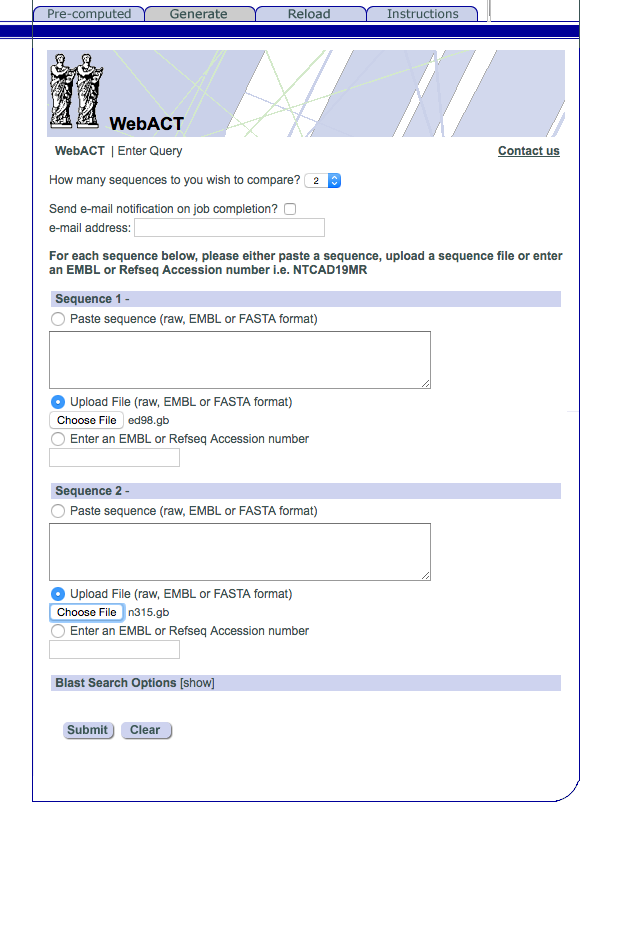
Now that we have the two Staphylococcus genomes we want to compare them. Upload them into webact
http://www.webact.org/WebACT/generate
Upload ED98.gbk at the top and N315.gbk at the bottom, like this:
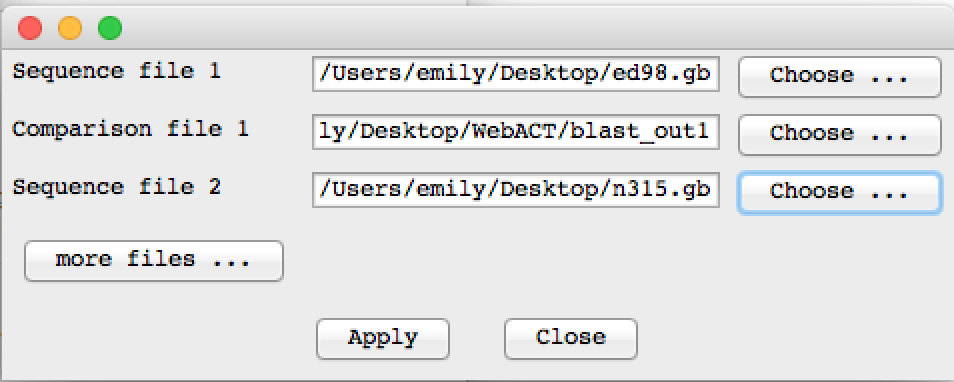
When this has finished download the results and open them in ACT. You can give the genbank files that you used to submit to webact and you use the blast_out1 as the comparison file.
You can get act here (and java):
- ACT should like something like this - can you find your mystery protein? hint: Try looking for the gene name or the locus_tag in the navigator
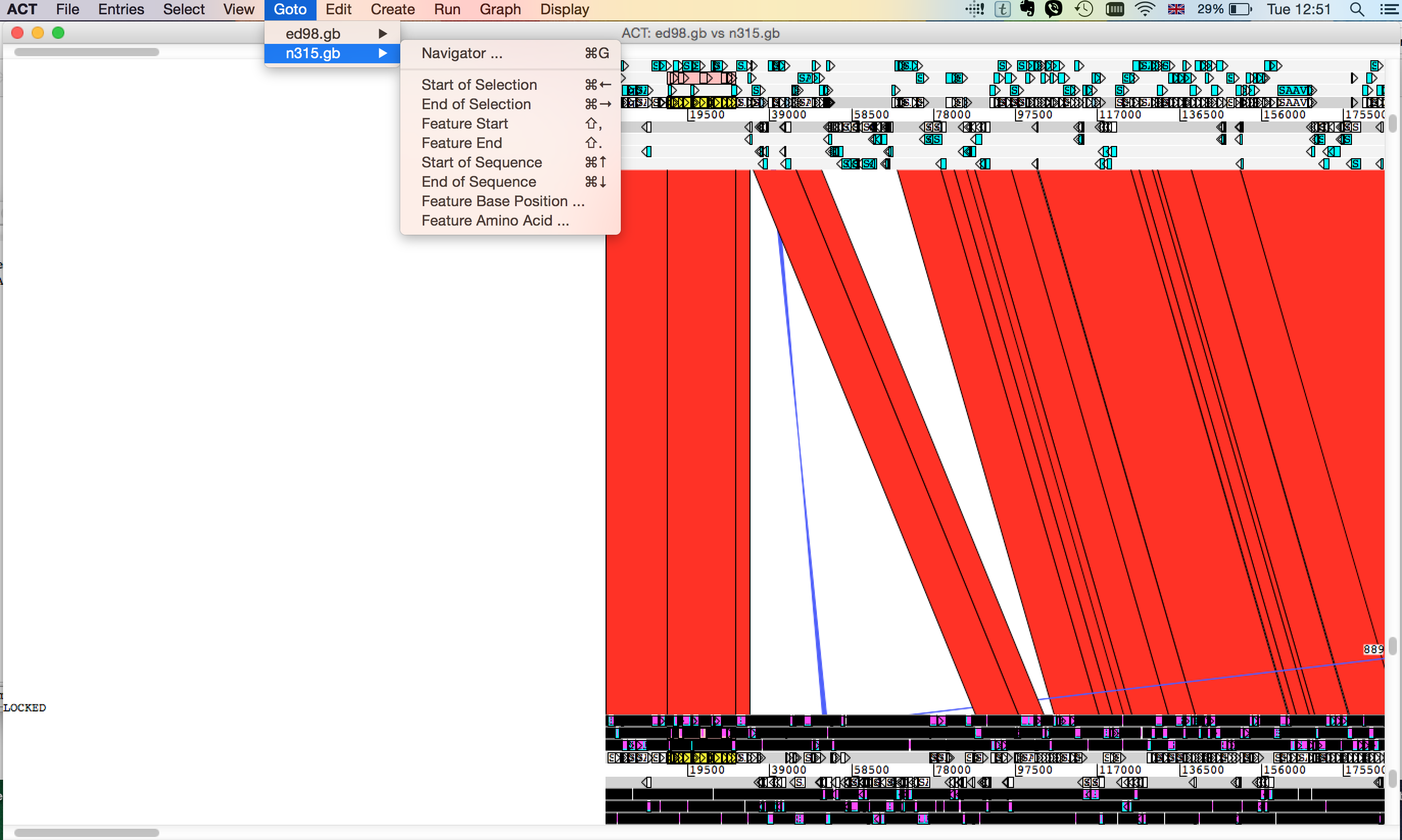

-
What do you observe about this region in relation to ED98?
-
Compare ED98 and N315 in these tools:
-
What’s the difference between the two strains.
-
In virulence - look up some of those genes, what can you infer from the presence of these genes? #clue - look up the host immune evasion proteins that got hits.
-
In antibiotic resistance?
-
Whats the difference between CGE and CARD. What’s the limitation with CGE versus CARD and vice-versa?
-
Can you download the .csv the CARD output? Then transform it to just show unique hits? #hint you did this in unix!
-
Do you think that the genomes are quite different?