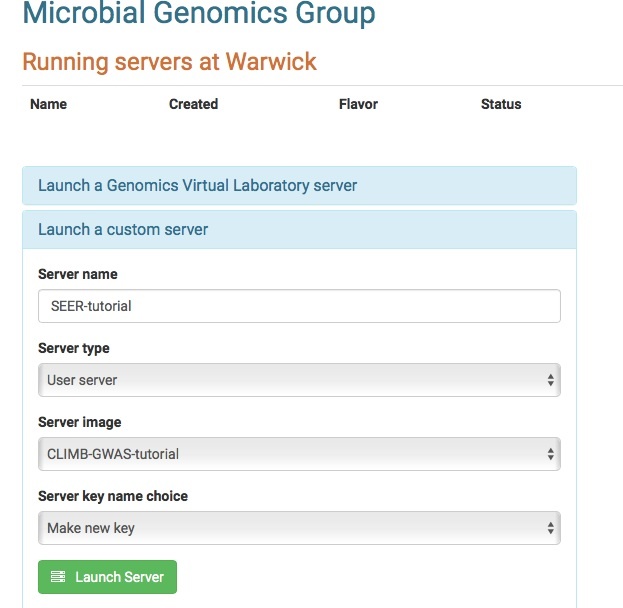
SEERS GWAS tutorial
John Lees and Sam Sheppard
Using 603 pneumococcal genomes isolated from the nasopharynx of US (MA) children. 224 were resistant or intermediately resistant to penicillin.
Croucher et. al. Nat Gen 6(45) 656–663 http://dx.doi.org/10.1038/ng.2625
We start with genomes assembled by velvet (in seer_data/assemblies) and the list of which were resistant and susceptible to penicillin (seer_data/penicillin.pheno)
- counts kmers
(not run)
fsm-lite -v -l fsm_files.txt -t tmp_idx -s 10 -S 593 > fsm_kmers.txt
split -d -n l/16 fsm_kmers.txt fsm_out
gzip fsm_out*
These first steps count the kmers from the assemblies and split the output into files to facilitate parallelisation later. We use kmers to test for association as these allow us to look for both gene and SNP variation simultaneously, and they don’t rely on a single reference genome.
As this step takes about 4 hours the results have been pre-calculated in seer_data/kmers
- correct for population structure, weighted by sequence identity of isolate genomes (without the use of a tree).
(population structure)
cd seer_data/population_structure
mash sketch -o reference …/assemblies/*.fa
mash dist reference.msh reference.msh > mash_distances.txt
perl ~/seer/scripts/mash2matrix.pl mash_distances.txt > all_distances.csv
perl ~/seer/scripts/R_mds.pl -d all_distances.csv -p …/penicillin.pheno -o projection
- Create a minhash of all the assemblies
- Calculate the pairwise distance between all samples
- Convert the mash output into a distance matrix
- Project the 603x603 distance matrix into 3 dimensions
Bacteria are clonal, and so a simple test of association will bring up all variants in a lineage associated with the phenotype (penicillin resistance) as well as the causal variants (SNPs in PVP genes). By assessing the similarity between samples we can approximate this population structure, and control for it in seer.
- Run all regressions kmer presence/absence Vs Phenotype
(seer)
cd …/run_seer
seer -k …/kmers/fsm_out00.gz -p …/penicillin.pheno --struct …/population_structure/projection --threads 8 --maf 0.05 > seer00.txt
This runs the association. It uses the counted kmers -k, the phenotype -p and the population structure calculated above --struct. We also filter out low frequency kmers, and multithread on the vm for speed.
This command can be run in a reasonable amount of time (<20 minutes) to get the output from one of the kmer files. Using less -S, have a look in the output file to see what seer generates as output.
This can then be filtered to just those kmers positively associated with resistance
filter_seer -k seer00.txt --pos_beta > seer00_filtered.txt
(or use answers in demo: filter_seer –k answers/00/stout –pos_beta
(not run)
parallel --results answers -j 8 seer -k …/kmers/fsm_out{}.gz -p …/penicillin.pheno --struct …/population_structure/answers/projection --maf 0.05 ::: 00 01 02 03 04 05 06 07 08 09 10 11 12 13 14 15
for i in $(find . -name stdout); do filter_seer -k $i --pos_beta | sed ‘1d’ >> seer_filtered.txt; done
This runs all the seer jobs in parallel on the climb vm and then filters them. This takes about two hours so results are provided in answers/seer_filtered.txt
(downstream)
cd …/downstream
~/seer/scripts/hits_to_fastq.pl -k …/run_seer/answers/seer_filtered.txt -b 10e-8 > seer_filtered.fastq
bowtie2 -p 8 --reorder -q -U seer_filtered.fastq --ignore-quals -D 24 -R 3 -N 0 -L 7 -i S,1,0.50 -x Streptococcus_pneumoniae_ATCC_700669_v1 -S seer_filtered.sam
~/seer/scripts/mapping_to_phandango.pl -s seer_filtered.sam -k …/run_seer/answers/seer_filtered.txt -m 20 > climb_phandango.plot
Less climb_phandango.plot
- Converts significant kmers to a fastq for mapping
- Maps significant kmers to a reference genome
- Visualise as a Manhattan plot in phandango
Note the message from bowtie2 -> if kmers are unmapped another reference can easily be mapped to without repeating any of the long steps above. Therefore the pan-genome has been tested, not just the core
- on local machine
(visualisation - local)
Copy the gff and plot file locally and drag onto http://jameshadfield.github.io/phandango/
scp ubuntu@137.205.69.99:~/seer_data/downstream/climb_phandango.plot Desktop
scp ubuntu@137.205.69.99:~/seer_data/downstream/Streptococcus_pneumoniae_ATCC_700669_v1.gff Desktop
The four biggest peaks respectively are pbp2x, orf15 (in the ICE), pbp1a, pbp2b
All three penicillin binding proteins!
Bonus question for fans of the pneumococcus: why does the ICE appear to be associated with penicillin resistance?