EDGE
The tutorials below illustrate some of the basic methodologies that can be employed for WGS analysis (assembly and mapping).
In order to get you familiar with bioinformatics WGS data we are going to use EDGE.
EDGE is an integrated genomics environment for population genomics, metagenomics and 16S analysis.
A server running EDGE is hosted on CLIMB at http://edge.climb.ac.uk
1: Example Population Genomics dataset - Carriage of S. aureus from Monkeys in the Gambia.
During this session we will show you how to upload, assemble and map an example fastq file. This will take some time to run.
We will go through an example, pre-computed dataset while the example isolate is being processed. During the afternoon session we will analyse the output of the run.
The genomes in this section were taken from the publication - “Transmission of Staphylococcus aureus from Humans to Green Monkeys in The Gambia as Revealed by Whole-Genome Sequencing.” (http://aem.asm.org/content/82/19/5910.long1)
Study Aims
Staphylococcus aureus is a gram-positive, commensal bacteria which is also an opportunist pathogen. S. aureus is a problem pathogen in the hospital setting, due largely to its acquisition of broad resistance to antibiotics. In this study S. aureus isolates were collected from both rural and urban Green Vervet monkeys from the Gambia. Samples were also collected from human infections in hospitals from the same region. A number of questions were posed including, is there evidence for human to monkey transmission or vice-versa, was the S. aureus infecting monkeys distinct from human isolates and was there any evidence of antibiotic resistance in monkey associated S. aureus.
Data
Five genomes (F8, F12, H2, H8 and H10), one from each of the sub clades of the novel monkey-associated clade, were selected as representative samples. These were assembled and annotated. H10 was then assembled and annotated using EDGE and a phylogenetic tree was produced which included a number of S. aureus reference genomes from the NCBI database and including the 4 additional monkey-associated isolates.
Raw read files for H10 can be downloaded from the ENA - http://www.ebi.ac.uk/ena/data/view/ERX12859651
The other samples from the study can be found here - http://www.ebi.ac.uk/ena/data/view/PRJEB12419
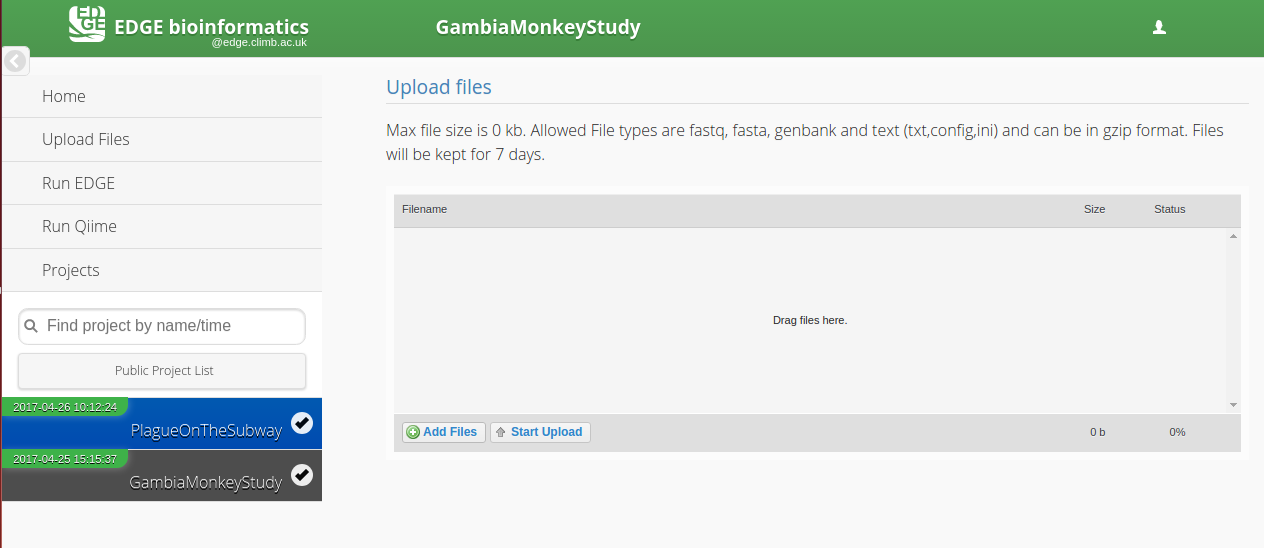
Uploading Files
Note: We will not be uploading/downloading files during this session due to the amount of time it will take to complete. We will be downloading a sample directly from the SRA in the next step.
Log-in to EDGE. If you have not made an account register by clicking on the user icon at the top of the screen and registering an account.
Click on the Upload files tab on the left hand side of the screen.
Either:
A) Drag and drop files onto the centre panel.
B) Click “Add Files” and select the appropriate files.
Click “Start Upload”
The progress bar and % indicator will give you feedback on how much of the task is completed.
Running EDGE
Click the “Run EDGE” tab on the left hand side of the screen.
This will bring up a number of drop-down boxes that contain separate steps in the workflow.
Input Raw Reads
Click on Input from NCBI Sequence Reads Archive(SRA) - click the YES box.
Input SRS2173782 to the SRA Accession box.
Note: Config files can also be used to reuse parameters that have been previously selected in “additional options”. You can also batch process multiple files using the same commands (see EDGE manual)
Fastq Files
File format:
Line 1 - record name; starts with ‘@’
Line 2 - DNA sequence
Line 3 - empty line; starts with ‘+’
Line 4 - phred-scaled quality scores
@cluster_2:UMI_ATTCCG
TTTCCGGGGCACATAATCTTCAGCCGGGCGC
+
9C;=;=9@48689:67AA<9>65<=>591
Sample Preprocessing
This section contains a number of read trimming and filtering options. Additionally if there is know contamination from a host/source then reads that map (align) to sources of contamination can be removed as this stage. Having filtered, high-quality reads with little to no contamination will produce better assemblies.
The majority of the default options should be suitable for most analyses, although the Trim Quality Level should be increased to 20.
Assembly
Assemblers are software that will take short or long NGS reads and attempt to reconstruct the underlying DNA sequence.
EDGE contains three assemblers: UDBA-UG, SPAdes and MEGAHIT. UDBA-UG and SPAdes are suitable for assembly of single samples. MEGAHIT is suitable for metagenomic assembly.
SPAdes is currently the most popular assembler for bacterial genomics. Select SPAdes in the assembler options.
Annotation
Automated CDS/ORF Annotation can be performed in two way using EDGE. Automated predication and annotation using PROKKA or transferring existing annotation from a reference genome using RATT. RATT would be suitable for studies looking at highly clonal isolates with a closely related reference genome. PROKKA is suitable for predicting ORFs in bacterial assemblies and provides good reference databases tailored to well-studied organisms.
Click on the Annotation drop-ddwon menu. Change the ‘minimum contig length for annotation option’ to 500bp.
Reference Based Analysis
Aligning of trimmed reads to one or more reference genomes is commonly used for calling high confidence variant sites. It is commonly referred to as ‘mapping’. Bases that are consistently different in reads relative to the reference genomes are called as SNPs.
EDGE supports two read aligners, Bowtie (default) and BWA-mem.
Contigs can also be aligned to the reference genomes. A low percentage of aligning contigs would indicate either contamination or a large amount of novel or divergent sequence.
- Click Reference Based Analysis On.
- Under ‘select reference genomes’ type in Staphylococcus aureus. Then click on S. aureus HO_5096_0412 and USA300_FPR3757.
Taxonomic Classification
A number of databases have been instituted in EDGE. These cover identification and classification of genus/species from read data (Kraken/BWA) or contigs (Kraken). There are also virus databases for viral samples and phage elements in bacterial samples (GOTTCHA-virus).
Phylogenetic Analysis
EDGE can produce phylogenetic trees of the sample of interest compared to preexisting datasets for a limited number of species. Alternatively, the user can select genomes from the NCBI or provide up to 5 genebank or fasta files. SNPs are then called by aligning the assemblies to a reference genome (designated by the user or randomly selected). These SNPs are then passed to either FASTTREE (quick and dirty) or RaxML (slow and clean) to produce a phylogenetic tree.
- Click on Reference analysis drop down ‘yes’.
- Click on ‘select reference genomes’.
- Select a number of reference genome to use to compute a phylogentic tree (e.g. select HO_5096_0412, USA300_FPR3757, JKD5007, Newman and ST239)
Specialty Gene Profiling
EDGE checks genes from samples against a number of different curated databases of genes that have been previously identified as being involved in virulence and antibiotic resistance. The trimmed reads and annotated ORFs can be check against these databases to identify genes of interest.
- Click ’ Speciality Gene Profiling’ - On.
Submit the Job
- At the bottom of the page click ‘Submit’.
- There should a green ‘successful submission’ message below the button.
- Job progress can be viewed in the Projects tab (left side of page) when you are logged in.
2: EDGE - is there anthrax on the subway?
While the S. aureus sample is running we will move onto a pre-computer example.
Choose
from the left menu.