Ancient DNA Metagenomics Tutorial
The skeleton of a 50- to 60-year-old male (skeleton 2568) was excavated from the abandoned medieval village of Geridu (Sorso, Sassari, Italy) in northwest Sardinia in December 1997. Thirty-two calcified nodules were found in the pelvic girdle, with diameters ranging from 0.6 by 0.7 cm to 2.2 by 1.6 cm (Fig. 1). A DNA extraction was performed on one of the nodules.
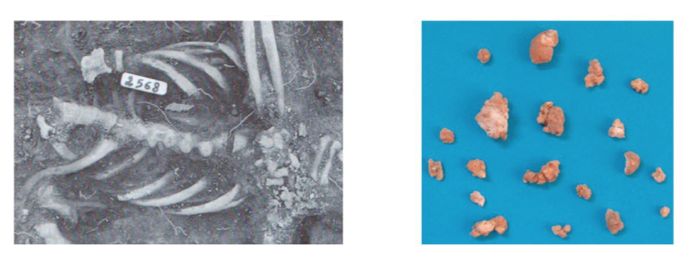
The nodule yielded 30 ng of DNA, which was used to construct a TruSeq Nano Illumina library. This library was sequenced on a single MiSeq run, which yielded just over 20 million paired-end sequences.
Login to VM
Login to your GVL server via SSH (or VNC in and use the Terminal).
If you don’t yet have a GVL server follow this tutorial first.
Create a new directory and move into it:
mkdir aDNA_tutorial
cd aDNA_tutorial
Download the Illumina FASTQ files from CLIMB CEPH data store (note how fast it is, you can use CEPH to store your own files):
wget http://s3.climb.ac.uk/tutorial-data/Nodule1_S1_L001_R1_001.fastq.gz
wget http://s3.climb.ac.uk/tutorial-data/Nodule1_S1_L001_R2_001.fastq.gz
List the files
ls
You will see the output files from the sequencing run:
Nodule1_S1_L001_R1_001.fastq.gz
Nodule1_S1_L001_R2_001.fastq.gz
We wish to know whether there are any bacterial sequences of interest in this nodule, so we run the program metaphlan. Metaphlan is available on the GVL via the homebrew-science repository. It was installed simply by running:
brew install metaphlan
Note how brew also installs the dependencies automagically, such as bowtie2!
For more details of the packages available on GVL see this forum post.
metaphlan.py --bowtie2db /mnt/gvl/apps/linuxbrew/.linuxbrew/Cellar/metaphlan/1.7.8/bowtie2db/mpa --nproc 8 --bt2_ps very-sensitive --input_type multifastq Nodule1_S1_L001_R1_001.fastq.gz nodule.txt
A few minutes later there will be an output file
nodule.txt
You can read this file in chunks by typing the following command:
more nodule.txt
Note the report of Brucella sequences in the file!
/mnt/gvl/apps/linuxbrew/Cellar/metaphlan/1.7.8/bowtie2db/
We wish to visualize the results in a graphical format
The metaphlan_hclust_heatmap.py script in the MetaPhlAn plotting_scripts folder can now be used to perform hierarchical clustering of both samples and clades to generate the heatmap. However, this script requires matplotlib and scipy to function so:
brew install matplotlib
brew install openblas
brew install scipy
mkdir output_images
/mnt/gvl/apps/linuxbrew/.linuxbrew/Cellar/metaphlan/1.7.8/plotting_scripts/metaphlan_hclust_heatmap.py -c bbcry --top 25 --minv 0.1 -s log --in nodule.txt --out abundance_heatmap.png
You can either download the png file from the server, or go via the interactive desktop, or via Jupyter notebook to see the output file.
Specifically, we are displaying the abundances for species only (default --tax_lev s), in logarithmic scale (-s log), reporting only the 25 most abundant clades (–top 25) according to the 90th percentile of the abundances in each clade (default --perc 90) with custom color map (-c bbcry). In this example, the clustering is performed with “average” linkage (default -m average), using “Bray-Curtis” distance for clades (default -d braycurtis) and “correlation” for samples (default -f correlation).
We now have convincing evidence that the sample contains sequences from the genus Brucella, but not which species.
We now wish to see which species of Brucella it resembles most closely, so we map the sequences from the nodule against reference genomes from the four species of Brucella that commonly infect humans: B. abortus, B. melitensis, B. suis and B. ovis.
You could find these reference genomes from the NCBI FTP site:
ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Brucella_abortus/
ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Brucella_melitensis/
ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Brucella_ovis/
ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Brucella_suis/
We run BWA to do this:
bwa index reference.fasta
bwa mem reference.fasta Nodule1_S1_L001_R1_001.fastq.gz Nodule1_S1_L001_R2_001.fastq.gz | samtools view -bS - | samtools sort - -o nodule.sorted.bam
Which reference does the most number of reads map to? (hint: use samtools stats)
Next we want to call out SNPs from our medieval Brucella genome, suggest using Torsten Seemann’s Snippy tool which is already installed on CLIMB: